Using the Two-Hybrid Screen in the Classroom Laboratory
Abstract
The National Science Foundation and others have made compelling arguments that research be incorporated into the learning of undergraduates. In response to these arguments, a two-hybrid research project was incorporated into a molecular biology course that contained both a lecture section and a laboratory section. The course was designed around specific goals for educational outcomes, including introducing research to a wide range of students, teaching students experimental design and data analysis, and enhancing understanding of course material. Additional goals included teaching students to search genomic databases, to access scientific articles, and to write a paper in scientific format. Graded events tested these goals, and a student evaluation indicated student perception of the project. According to our analysis of the data, the yeast two-hybrid screen was a success: several novel clones were identified; students met expectations on graded lab reports, the poster session, and the final paper; and evaluations indicated that students had achieved the outlined goals. Students indicated on the evaluations that the research project increased their interest in research and greatly improved understanding of the course material. Finally, several students in the course intend to submit the findings of the research project to an undergraduate research journal.
INTRODUCTION
The National Science Foundation and others have made compelling arguments for incorporating research into the education of undergraduate students (National Science Foundation, 1996; Rothman and Narum, 1999). They further recommend introducing research to students early in their college education. This research-oriented approach to classroom learning has been well documented (Layman, 1996) and tested in the undergraduate teaching laboratory (Henderson and Buising, 2001). We have incorporated a classwide research project into a 200-level lecture-and-laboratory course in molecular biology at Connecticut College. The design of learning experiences and student assessment was based on an integrated course design model (Fink, 2001). Connecticut College is a small liberal arts college with a history of encouraging undergraduate research in the sciences.
This laboratory project was designed to introduce scientific research to a broad range of students and to demonstrate to students the excitement of discovery. In addition, students are taught to design experiments and to analyze data, including the proper use of controls. The design of the project and that of the course were integrated so that the laboratory would enhance student understanding of the course material. Goals specific to cell biology were also identified: upon completion of the course, the learner will have gained the skills necessary to 1) search genomic databases, 2) search and electronically access scientific journal articles, and 3) write a paper in scientific format. These goals were not shared with the students so that the goals would not influence the students' evaluations. The achievement of these goals was assessed by graded events designed to test the goals and through analysis of student evaluations of the project.
This project would work well in a cell biology, molecular biology, or genetics course because the screen encompasses basic cellular processes, including transcription, translation, and DNA replication, and because the instructor chooses the cellular system to which the project is applied. The course is designed for one college semester with a 4-h laboratory period that meets once per week, although this project could be scheduled into a 3-h lab with only a few modifications. The project is appropriate for sophomores to seniors, it spanned 8 weeks of the course, and it included 12 students in each laboratory section.
The project involves a two-hybrid screen in yeast (Fields and Song, 1989). The screen includes many important laboratory techniques, including bacterial and yeast transformation, a cDNA library screen, isolation of plasmid DNA, restriction enzyme digestion, DNA sequencing, and utilization of genomic databases. This project allows students to observe results of cellular processes covered in the course curriculum. For example, coinciding with lectures on transcription, laboratory sessions allow students to observe yeast turning blue due to activated transcription of the lacZ gene. Thus, students experience a visible result of an abstract, molecular event. Throughout the project, students connect topics learned during the semester and often relate topics learned in lecture to the ongoing research in the laboratory. Examinations given in lecture indirectly supported the research project by incorporating problem-based learning questions. Each exam contained questions typical of problems that students might encounter in laboratory research. Because each student pursues his or her own results throughout the 8-week project, students become invested in the outcome of their experiments and are excited to see their results and to analyze their data.
In addition to the many benefits to students in the class, another advantage to screening a library is that the data obtained can be pursued by students as independent study or honors' thesis topics. The yeast two-hybrid screen can be used to search for unknown protein interactors of a protein of interest to the instructor; thus, this research can directly benefit ongoing research at the college or in the faculty member's lab. Students who are interested in pursuing work on a particular protein-protein interaction can continue work as an independent study. The potential topics for research following the screen have almost endless possibilities, from bioinformatics to bench science. For instance, students can pursue questions relating to protein binding in an in vitro system or use immunofluorescence to investigate the subcellular localization of the two proteins.
Finally, the excitement that students experience in performing original research enhances their interest in the course and in science. At the conclusion of the screen, students research the protein that they identified in the screen by accessing the scientific literature. Students then form hypotheses regarding the significance of the protein-protein interaction that they identified and even propose experiments to test their hypotheses. One student commented that he or she liked the “excitement of trying to discover new things.”
MATERIALS AND METHODS
The Two-Hybrid System
A yeast two-hybrid screen is a way to search for protein-protein interactions. The goal is to isolate a protein or proteins that interact with a “bait” protein; this bait protein can be almost any protein from any organism. The only requirement is that a cDNA library be obtained from the same or a closely related organism. A useful analogy compares this experiment to fishing. A bait is put into the yeast and the students “go fishing” to find “fish” or “prey” (proteins that interact with the bait). A cDNA library provides the possible prey. A good cDNA library will provide a DNA copy of each mRNA in a particular cell type. Excellent cDNA libraries can now be purchased from several companies, including Gibco products from Invitrogen Corp. (Carlsbad, CA; ProQuest #11386-018), the source used in this laboratory. This protein-protein interaction search is performed in yeast, which acts as the host for the library screen. The yeast contains two plasmids: one that contains the gene for the bait protein and one that contains the cDNA library. Once inside the yeast cell, each of these plasmids will express the genes as proteins. The system is constructed so that any protein encoded by the cDNA library that binds the bait will cause transcription of a reporter gene and essentially provide a “tug on the line” that lets the researcher know that he or she has a “hit” (a potential protein-protein interaction). Thus, this system provides a visual readout of transcription.
The transcription factor that activates the reporter gene is made up of two distinct fusion proteins that can combine to reconstitute transcriptional activity (Ma and Ptashne, 1988; Figure 1). The cDNA of the bait is fused to the coding sequence of the DNA-binding domain (DBD) of a transcription factor, and the cDNA library is fused to the sequence encoding the transcriptional-activation domain (TAD) of a transcriptional-activating protein. If and when these two hybrid proteins bind (the fish bites the bait), a transcriptional-activating factor is reconstituted, the two proteins bringing together the two essential parts: the TAD and the DBD. This reconstitued factor will activate transcription of only a reporter gene or reporter genes that are under control of that transcription factor (the Gal4 transcription factor and binding sites are used here; Figure 2). The name of the system, two-hybrid, stems from the fact that the bait fused to the DBD and the cDNA fused to the TAD form two hybrid coding sequences and proteins. This system was first described by Fields and Song in 1989 and has been improved over the years to allow more efficient screening of baits (Finley and Brent, 1994; Grossel et al., 1999; Hanes and Brent, 1989; Inouye et al., 1997; Vidal et al., 1996a). Detailed protocols for yeast two-hybrid screening are available in several manuals (Ausubel et al., 1999; Burke et al., 2000).
Our screen used full-length cyclin-dependent kinase (cdk) 6 as bait in each of the three screens we performed. This bait was chosen on the basis of the instructor's research interests. Any bait can be used as long as a cDNA library from the same or a similar organism is available to screen. Full-length clones are recommended because using the full protein not only increases the probability that the protein will fold correctly, but also increases the number of protein domains available for interaction with another protein. Yeast strain HW18 (Genotype: MATa ura3-52 leu2-3 112trp1-901 his3Δ200 ade2-101 gal4Δ gal80Δ can1R cyh2R Gal1::HIS3@LYS2 GAL1::lacZ SPEX4::URA3) containing plasmid pGalcdk6 was used for this screen and is available upon request. The two-hybrid screen is also available as a kit from several companies, including Invitrogen (ProQuest series 10835).
The Laboratory Schedule
The 8 weeks of this project consist of 6 weeks of laboratory work and 2 weeks of DNA sequencing and computer database searching. In the first week of the project, yeast that retain the bait plasmid are transformed with the cDNA library plasmid. Each plasmid contains a gene involved in the production of an amino acid (Figure 2B), so that a process similar to antibiotic selection in bacteria can be used in selecting the yeast that were successfully transformed. After selection for yeast that contain the cDNA library, the screen is performed. Yeast colonies are transferred by a simple method, called replica-plating, onto medium that selects for yeast that contain a positive protein-protein interaction. The primary screen tests for transcriptional activation of the HIS3 reporter gene as determined by the ability to grow on medium lacking histidine and containing 3-amino-triazole (3AT), which increases the stringency compared with that of medium simply lacking histidine (which thus requires the yeast to produce more histidine in order to survive). In the second week, potential positive yeast (those that test positive in the first round and contain potential interactors) are retested for growth on plates lacking histidine and tested for expression of a second reporter gene, lacZ. In Vidal's system (Vidal et al., 1996a, 1996b, 1998), both reporter genes (HIS3 and lacZ) are under control of binding sites for the Gal4 transcription factor (Figures 2A and 3). Expression of the lacZ gene is indicated by blue color formation in yeast in aβ -galactosidase assay (Figure 3). Controls are necessary to ensure reliable results of these assays. In our screen, three controls are used: a strong positive (Fos/Jun), a weak positive (pRB/E2F), and a negative control (empty vector). (See Appendix, Experimental Details and Protocols, for details.) One strength of this particular yeast system with two reporters is that it decreases the likelihood of identifying false-positives (Vidal et al., 1996b; Gibco kit). All hits isolated thus far in this screen appear to be true cdk6 interactors, but as with all two-hybrid hits, interactors need to be confirmed by in vitro binding studies. Controls for false-positives due to nonspecific activation of reporters (e.g., by the bait alone) are not included in our lab, but they can be included or made part of independent studies by students who want to continue research after the project is completed.
In this screen, the yeast host is used to select a handful of protein-protein interaction events from a cDNA library representing products of as many as 30,000 genes. This fact seems remarkable to students. Each student screens thousands of events, from which some students identify one or more protein-protein interactions out of thousands of protein combinations. The process of transcription in a yeast host allows the student to find a needle in a haystack, an important lesson about designing good biological screens.
Figure 1. Graphic representation of the yeast two-hybrid system. The two-hybrid system is based on the property of transcription factors that allows each domain to work independently of the other. (A) The DNA-binding domain (DBD) binds DNA, and the transcriptional-activation domain (TAD) stimulates the promoter complex. (B) In the two-hybrid system, the DBD and the TAD are separated and can be reconstituted only by a protein-protein interaction between a bait protein and a prey protein. This protein-protein interaction allows activated transcription of a reporter gene. In this diagram, the lacZ gene will give a phenotypic readout of blue color formation in yeast.
It is important to note that not every student isolates a hit, but after the initial screen, the class works as a group and at each step focuses its labors on the positive outcomes that were isolated by some members of the class. The number of clones tested (yeast cells transformed) varies from 3,000 to 12,000 per student. The number of hits is directly related to the number of clones tested and varies with the bait used. In the third year of this screen, with 43 students, 190,000 total clones were screened, five independent hits were isolated, some isolated more than once. Two hits were known interactors, cyclin D3 and p19, and two were novel interactors. One hit that did not match a known gene in the databases was also isolated; all others were mapped to known open reading frames. At least one potential hit was lost at some point throughout the DNA-isolation procedure. To rescue lost hits, the instructor can intervene during or after the project.
In the third week, using only the yeast that tested positive for interactions, the class isolated the plasmids from the yeast chromosomes and yeast proteins. The procedure used to purify DNA from yeast is called a“ smash and grab” and is conducted during two lab sessions (weeks 3 and 4). In essence, a smash and grab is a plasmid prep in yeast. The yeast cells are lysed, the proteins are denatured, and plasmid and genomic DNA is isolated (Ausubel et al., 1999; Sambrook et al., 1989). This DNA is then transformed into competent bacteria (Sambrook et al., 1989). Bacterial transformation allows the plasmids to be amplified for restriction endonuclease analysis and sequencing. Although either of the two plasmids (bait or prey) will transform bacteria equally well, the yeast can be enriched for the cDNA library plasmid prior to the smash and grab by using auxotrophic selection in the yeast culture medium. Once transformed, bacteria replicate the plasmids, which provides sufficient quantity to allow sequencing of the cDNA inserts. In week 5, the plasmid DNA is purified by using Qiagen columns (Qiagen Inc., Valencia, CA) and digested to confirm the presence of a cDNA insert. In week 6, the DNA is sequenced.
In our lab, Pfizer Inc. (Groton, CT) provides us a tour of their sequencing facility, and students learn about high-speed sequencers used in the human genome project. However, a laboratory session devoted to sequencing could easily be added and would likely take 2 weeks to complete. Sending DNA to a university facility to be sequenced is also an inexpensive and fast alternative ($15-$20 per reaction). After sequencing, the student has the sequence of the unknown cDNA that encodes a protein that interacted with the bait protein to activate transcription in yeast. In the seventh week, a search tool called BLAST (Altschul et al., 1997) is used in an attempt to identify the cDNA clone. The search can be performed on any computer with Internet access and returns a list of genes with sequences similar to the sequence of the cDNA identified by the students. The information returned gives several best matches to the sequence entered (see Ausubel et al., 1999). The sequence of the cDNAs usually allows the student to determine which protein the sequence encodes. Having learned the identity of the interacting protein, students perform literature searches to understand the function of the protein, often making important connections to information they learned previously.
Figure 2. Graphic representation of the reporter genes and plasmids used in the two-hybrid screen shown in Figure 1B. The screen reported here includes two reporters and two plasmids. (A) A reconstituted transcription factor will activate transcription of the HIS3 gene and simultaneously activate transcription of the lacZ gene. The HIS3 reporter can be selected for growth on plates containing 3-amino-triazole (3AT), and the lacZ reporter can be assayed by performing a β-galactosidase assay that will cause blue color formation in yeast. (B) The plasmids transformed into the yeast for the two-hybrid screen. The Galcdk6 plasmid encodes the Gal4 DBD fused to cdk6. This plasmid also encodes the LEU2 gene for use in auxotrophic selection. The second plasmid encodes the cDNA library fused to a sequence encoding a TAD. This plasmid also encodes the marker TRP1, a gene involved in tryptophan biosynthesis.
The final week of the project is spent discussing the hits. Our experience indicates that this is best done as a poster session in which students can exchange the knowledge they have gained with their classmates. In this poster session, posters were presented by groups of 3 to 6 students who all shared a common hit. This year's class of 43 students in four lab sections resulted in eight posters representing the four hits that were isolated. The overlap of groups presenting the same hit is helpful to students because individual groups commonly form distinct hypotheses regarding the implication of the bait-prey interaction. The poster session allows students to analyze their work and to connect their findings with knowledge gained in other parts of this course.
RESULTS
A yeast two-hybrid screen has been conducted as part of a molecular biology course for 3 consecutive years. cDNA libraries derived from different tissues have been screened with the same bait (full-length cdk6) chosen on the basis of the interest of the faculty member teaching the laboratory (Hunter and Pines, 1994; Morgan, 1995). A human fetal brain library was used for 2 yr, and a human adult brain library was used the third year. Assessment of the effectiveness of the project has been performed through graded events and through analysis of student evaluations of the project goals (as defined by instructors at the start of the term). Three of the goals of the project were met through the integrated course design. Specifically, a broad range of students was introduced to scientific research, and the project demonstrated the excitement of discovery to the students. In addition, students were taught how to access genomic databases by individual instruction, which ensured that each student acquired the skills needed to search these databases.
Figure 3. Yeast reporter plates showing a typical outcome of a yeast two-hybrid screen. Controls for the experiment included a strong positive (Fos/Jun; ++), a weak positive (pRB/E2F; +), and a negative (empty vector; -) control yeast strain. Circled yeast colonies are hits. (A) The master plate containing transformed yeast colonies that are suspected hits and controls (++, +, -) for the experiment. (B) A 3AT reporter plate. Growth indicates transcriptional activation of the HIS3 gene. Circled yeast are hits. (C) Aβ -galactosidase assay. Blue color formation indicates transcriptional activation of the lacZ gene and a hit. Note that not all yeast that grew well on the 3AT plates turned blue, which likely indicates a weaker protein-protein interaction.
Graded events relating to the research project included a laboratory report that was written in the format of a scientific paper, a presentation of a poster at a poster session, and a final paper based on the research project. During the semester, students wrote three lab reports in scientific format. The course instructors graded each report, and detailed feedback was provided to enhance student understanding of the format used. The first two lab reports were complete reports written on separate laboratory exercises. The first was based on a 2-week polymerase chain reaction (PCR) experiment, and the second report described the results of a 2-week cloning experiment. These exercises were chosen in part to provide skills that would assist students in the two-hybrid screen and in part because of their inherent importance in the field. In the first lab report, many students had difficulty writing in a detailed and concise manner and had difficulty distinguishing differences between sections of a research paper (e.g., the Materials and Methods section and the Results section). By the second attempt, students were able to produce reports in proper scientific format. The students' progress was evident in the trend of better grades on the second and third reports. This year, the average grade on the first lab report was a B-, the average grade on the second report had improved to a B+, and this average improved slightly on the final two-hybrid report. The grading on all three reports was consistent, with careful attention paid to inclusion of detail, to concise writing, and to careful analysis of the data. The instructors' impression that the students had learned the scientific format by the second report is consistent with the grade trend. (See Appendix, Sample Two-Hybrid Project Lab Report, for an example.)
Students were also graded on the presentation of their posters. Grading was based on the students' ability to communicate results, to answer questions regarding the controls used, and to understand a biological screen. All students demonstrated excellent understanding of the experimental controls used and their importance to the interpretation of data. The lowest score earned on the poster presentation was a B+, and the class average was an A-, which indicates that students met or exceeded our expectations in understanding the scientific method used in the project, including the proper use of controls. (See Appendix, Sample Two-Hybrid Project Posters, for examples.)
Objectives were directly measured by the evaluation of a final paper. Students wrote a paper on what is known about the function of a protein isolated in the screen and formed a hypothesis of how this function may be affecting the function of the bait protein with which they screened the library. This type of examination incorporates knowledge gained in the class and allows the student to apply his or her creativity and self-expression to the formation of the hypothesis. This paper was comprehensive and challenging and required students to accomplish the goals set out at the beginning of the course. For example, to write the exam, the learner had to perform literature searches, read relevant scientific articles, and summarize the functional properties of the protein. The reference sections of these final exams were analyzed quantitatively and qualitatively. The quantitative analysis indicated that 4 students reported three or fewer primary journal articles, 31 students reported between four and nine articles, and 3 students cited nine or more scientific articles. The qualitative analysis indicated that 33 students referenced articles that were categorized as outstanding. This category required references to be relevant to the paper topic and published in high-impact journals. The remaining 5 students cited articles categorized as excellent, a rating indicating that the articles were relevant but were published in journals with less scientific impact. Because all students completed the examination and because journal searching was a required part of that examination, the results clearly indicated that every student met the goal of searching and accessing scientific journal articles.
The project was also assessed through analysis of student evaluations completed at the end of each of the 3 yr the screen has been conducted. Each year, students completed a general evaluation that assessed interest in the course and the learners' ability to apply the laboratory experience to the course material. In the third year, a more detailed assessment was performed upon completion of the course, and the results are presented here. The evaluations were designed to assess students' perception of the outlined goals. Thirty-eight students completed the evaluations. In the evaluation, students were asked to rate their lab skills before and after the two-hybrid project on a scale of 1 to 5, with 1 being “great” and 5 being“ poor.” Twelve students rated their skills before the experiment high (1 or 2), whereas 36 students rated their skills after the project high. This finding indicates that 92% of the students who rated their skills less than high now rate them high. In response to a separate question, 15 students rated their interest in scientific research a 1 or a 2 before the two-hybrid project, whereas after the project, 30 students indicated their interest in research at a level of 1 or 2. This finding represents a twofold increase in the number of students interested in scientific research after the two-hybrid yeast screen. Although 5 students indicated that the project did not influence their likelihood of pursuing research, it is possible that these students already knew that they wanted to pursue research.
Data from several questions regarding interest in scientific research, student understanding, and the impact on understanding course material are presented in Table 1. These questions addressed the broad-based goals of the laboratory, including the effect of the project on student interest in the course and in research, and the influence of the project on understanding course material. As shown in Table 1, 18 students (47% of respondents) ranked this laboratory as the best lab they had taken at Connecticut College. This response is remarkable considering that 81% of the students in this course were juniors or seniors. Also, 36 of 38 respondents indicated with a 1 or a 2 that the two-hybrid project influenced their understanding of course material. Thirty-four students indicated with a 1 or a 2 that the lab report contributed to their understanding of the research project.
| Answersa | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| YES | NO | ||||||||
| Question | 1 | 2 | 3 | 4 | 5 | ||||
| Did the THPb increase your interest in the course or in molecular biology? | 20 | 11 | 2 | 1 | 3 | ||||
| Has the THP influenced the likelihood that you will pursue research outside the classroom? | 14 | 11 | 6 | 2 | 5 | ||||
| Did the THP influence your understanding of course material (transcription, sequencing, cloning, etc.)? | 24 | 12 | 2 | 0 | 0 | ||||
| Did the poster session help increase your understanding of the project? | 9 | 16 | 8 | 5 | 0 | ||||
| Did the lab report help increase your understanding of the project? | 20 | 14 | 2 | 0 | 1 | ||||
| BEST | WORST | ||||||||
| 1 | 2 | 3 | 4 | 5 | |||||
| How did this laboratory compare with other lab courses you have taken at Connecticut College? | 18 | 15 | 4 | 1 | 0 | ||||
Goals that were specific to cell or molecular biology were also assessed in this evaluation, and the results are presented in Table 2. The questions in this section measured an increase in understanding of particular subject areas. The scale ranged from 1 (increased understanding) to 5 (no increased understanding). The evaluation strongly indicated that students believed that they gained a greater understanding of proper use of controls (32 students chose a 1 or a 2) after completing the project. Students also indicated an increase in understanding genomic databases (35 students answered with a 1 or a 2) after completing this project. Although students were less positive about the impact of the project on their literature-searching skills (26 chose a 1 or a 2), several students stated that they already had those skills when they entered the course.
| Answersa | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| YES | NO | |||||||||
| Subject area | 1 | 2 | 3 | 4 | 5 | |||||
| Question: Did the lab increase your understanding of any of the following? | ||||||||||
| Proper use of controls | 16 | 16 | 3 | 1 | 2 | |||||
| The human genome databases | 20 | 15 | 4 | 0 | 1 | |||||
| Literature searching–electronic and print | 13 | 13 | 8 | 1 | 4 | |||||
| How to write a scientific paper | 15 | 15 | 5 | 1 | 2 | |||||
| How to read a scientific paper | 6 | 19 | 9 | 5 | 3 | |||||
| How to analyze data | 10 | 15 | 6 | 2 | 2 | |||||
| Responsibility for a project | 24 | 6 | 5 | 1 | 2 | |||||
Open-ended questions were also asked to determine the overall impact of the research project. The students were asked, “What did you like about doing an original research project as part of your lab class?” and“ What didn't you like?” Several students agreed on what they did not like about the project, commenting on the “ups and downs” of the project, the long labs, and frustration experienced with the lack of their individual results. A common response to the question of what students liked was the “excitement of trying to discover new things” and“ gaining experience and skills,” as well as “pride in work.” Several students also agreed that they enjoyed working with others on the lab and especially on the poster presentation. One student commented, “It (research) isn't just working in a secluded lab all alone.” In response to a question addressing why students liked or disliked the lab, one student commented, “Every part of the lab was related to lecture. We were able to apply what we learned.”
DISCUSSION
This two-hybrid research project has been incorporated into the molecular biology course at Connecticut College for the past 3 yr. Each of those years, students were positive in their evaluations of the laboratory, often stating that they enjoyed the break from doing weekly “canned” labs in which the outcome was known and they were expected to go through the motions of the experiment anyway. Such statements highlight a source of boredom and frustration with conventional laboratory classes for the students who often miss the fact that they are learning valuable techniques through standard lab exercises. The value of a research project is that the learner gains not only skills but also the opportunity to experience the excitement of discovery. Many students stated that they are more invested in the outcome of their work when they think they are responsible for a long-term project and when they are involved in “real-world” research. Several students have commented that they prefer this approach because it helped them understand course material presented in lecture. One student commented, on the project evaluation, “It (the research project) really helps cement the ideas from the lecture and it made me want to really learn the lecture material rather than just learn it for the course.” On the basis of the outcomes of graded events and analysis of course evaluations, our assessment of the course is that it has met or exceeded the goals with which we began the project. One of the major goals of the project was to introduce students to the excitement of discovery. Clearly, the twofold increase in student interest in research after the course indicates that exposure to research in the classroom setting can spark student interest.
Several of these goals were also assessed through a graded lab report, a poster session, and a final paper. Student performance on these events met the goals and exceeded our expectations. Students at the poster session demonstrated the ability to analyze their results and an understanding of the proper use of controls. Students wrote lab reports in clear, concise language, displaying an excellent understanding of the scientific format. The final paper clearly demonstrated that all students were able to research a scientific topic and produce relevant scientific references while distinguishing the quality of articles produced in the search (see Appendix, Sample Two-Hybrid Project Final Paper, for examples).
Several students intend to submit the findings of their classroom research to an undergraduate research journal. This experience will help students to understand the process of writing and submitting work for peer-reviewed journals and will likely further enhance their interest in research. Students who have participated in research-oriented learning and have continued their studies at the graduate level or have pursued research-oriented work have commented that they were better prepared because of this experience (C. Halsey, S. Sheehan, and D. Krull, personal communication).
From the faculty member's perspective, the weakness of this project is that there is considerable time involved in setting up the experiment. Many plates need to be prepared, and a cDNA library needs to be obtained. These cumbersome tasks could be alleviated if a kit would become available that would provide the cDNA library, selection and reporter plates, competent bacteria for transformation, and perhaps even access to sequencing. Regardless, the bait needs to be cloned into the plasmid with care to ensure a proper reading frame, and it is important to confirm by immunoblot that the protein is made (by Western blotting) before the screen is started. If this project can be of benefit to the faculty member, it is at least not only preparation for the course but also an investment in the faculty member's research program.
This year, the laboratory was concluded with a poster session. This session allowed students not only to present work on their own hit, but also to learn about the other proteins cloned by classmates. Those who pursued the same protein were able to compare references and information that they discovered, while discussions with groups that pursued separate leads allowed students to form a more complete picture of the role that the bait protein may play in the biology of the cell. At the poster session, students were engaged and inquisitive, eager to learn from their colleagues as well as the professors.
In conclusion, this project was highly successful, stimulating student interest in class material and in scientific research. One student stated in her lab report, “This experiment most definitely was successful in pulling all that we had learned in the semester back together. When the experiment was started it was a little unclear how this related to the material of the course, but as the weeks went on it became more and more clear. It was very interesting to be able to take part in this experiment as a way to synthesize a course. I enjoyed learning how research works and I think the fact that it was all hands on helped me grasp and explain the concepts of the course better than I could have from just reading or lecture.”
APPENDIX
EXPERIMENTAL DETAILS AND PROTOCOLS
Yeast Strain
Yeast strain HW18 (MATa ura3-52 leu2-3 112trp1-901 his3Δ200 ade2-101 gal4Δ gal80Δ can1R cyh2R Gal1::HIS3@LYS2 GAL1::lacZ SPEX4::URA3) was used for the classroom two-hybrid screen. HW18 is derived from MaV52 (Vidal et al., 1996) and was constructed to allow differential screening using multiple baits (Grossel et al., 1999). HW18 has three reporter genes—two of which depend on Gal4 binding sites in their promoters (HIS3 and lacZ) and one, URA3, dependent on LEXA binding sites in the promoter regions (Grossel et al., 1999). Thus, HW18 allows screening of two different proteins or“ baits” fused to two different DBDs (DNA-binding domains), each bait activating heterologous (Gal4-driven or LEXA-driven) reporter genes. In the first 2 yr of the classroom two-hybrid screen, the library was screened with two baits: LEXA-cdk4 and Gal4-cdk6. As a way to make the system more accessible to undergraduates, the library was screened with only the cdk6 bait in the third year. Simplifying the system greatly helped with student comprehension. In strain HW18, the two Gal4-driven promoters (HIS3 and lacZ) each contain five Gal4 binding sites upstream of the start site of transcription. The Gal4-dependent yeast two-hybrid system is available as a Gibco kit from Invitrogen (ProQuest series 10835).
Yeast strain HW18 was transformed with the Gal4-cdk6 expression plasmid, pGalcdk6. Plasmid pGalcdk6 encodes a fusion protein of the Gal4 DBD in the reading frame with the full-length cDNA of human cdk6. This plasmid also contains the LEU2 marker for auxotrophic selection, an ampicillin resistance gene for selection in bacteria, and an ARS/CEN origin of replication. Immunoblots of transformed HW18 confirmed expression of the cdk6 fusion protein. This transformed yeast strain, HW18/pGalcdk6, retains the plasmid when it is grown under auxotrophic selection (synthetic complete media lacking leucine, SC-L) or stored at -80°C in 15% glycerol.
Yeast Transformation
Seven to 14 d prior to transformation, yeast strain HW18/pGalcdk6 was streaked onto SC-L; plate preparation protocol is outlined below. Yeast grow to full-size colonies in about 3 d, are stable on a plate stored at 4°C for 1 to 2 weeks, and can be continually passaged onto fresh SC-L for at least several months. Yeast should be maintained at room temperature (RT) throughout the transformation protocol. The transformation should be monitored by using no DNA as a negative control, and transformation efficiency can be determined by counting colonies grown on a selective plate (SC media lacking leucine and tryptophan, SC-L-T). Proper sterile technique throughout the protocol is necessary to prevent contamination. Preparations of media, plates, and solutions are outlined below. During the screening of the yeast and the isolation of yeast containing potential interactors, students need to attend lab several times a week (for approximately 1 week). This extra time spent in lab is more than made up for in the very short lab session in which students prepare their master plate from their screened yeast. In our experience, students enjoy checking up on their screen and often require help in choosing which yeast contain potential hits.
The following protocols are formulated for 12 students (one lab section in our course):
Pre-lab preparation by the instructor:
One day prior to transformation, densely streak two SC-L plates with a heavy inoculum of HW18/pGalcdk6 (or another yeast strain) from an actively growing SC-L plate, and incubate at 30°C overnight.
Seven hours prior to transformation, suspend yeast cells in 650 ml of RT YPD/Glucose media in a 1- to 2-l flask until the OD600 is 0.13-0.15. We time this to be coordinated with our lab and notify the students that the lab may run long that day. Dogma dictates that yeast need to be transformed immediately and not stored prior to use, although the possibility of storing competent yeast on ice until it is ready to use could easily be experimentally determined. Recently, high-efficiency competent yeast have become commercially available (Gibco Life Sciences), and this yeast may provide a more time-sensitive method for transformation.
Incubate the culture with vigorous shaking at 30°C for 5 to 7 h until the OD600 is 0.3. The efficiency of transformation is greatly affected by the growth stage at which yeast are transformed. For the most efficient transformation, harvest yeast at exactly OD600 = 0.3.
Each student:
Using sterile technique, transfer 50 ml of yeast at OD600 = 0.3 into a 50-ml tube. Centrifuge the tube in a tabletop centrifuge at 1800 rpm at 20°C for 5 min.
Pour off the supernatant. Using sterile technique, resuspend the yeast pellet in 20 ml sterile water.
Centrifuge the tube at 1800 rpm at 20°C for 5 min.
Pour off the supernatant and resuspend the yeast pellet in 4.5 ml freshly made TE/LiAc solution from stock solutions of Tris, EDTA (ethylenediamine tetraacetic acid), LiAc (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 100 mM LiAcetate).
Centrifuge at 1800 rpm at 20°C for 5 min.
Pour off the supernatant and resuspend the yeast pellet in 250 μl fresh TE/LiAc solution.
Into each of four labeled microcentrifuge tubes, add the following:
100 μl yeast from step 6.
1 μg cDNA library plasmid (Gibco, ProQuest #11386-018; three tubes) or no DNA (one tube for a negative control).
5 μl of 10 mg/ml denatured salmon testes DNA (Sigma Chemical Co., St. Louis, MO; D7656). To denature, boil for 3 to 5 min, then quick-cool in an ice-water bath.
600 μl freshly made PEG (polyethylene glycol) solution from stocks of PEG, Tris, EDTA, LiAc (40% PEG [Sigma Chemical, P3640], 10 mM Tris-HCL, 1 mM EDTA, 100 mM LiAc). Add PEG solution last, then mix well by vortexing.
Incubate all four tubes at 30°C for 30 min.
Add 70 μl sterile DMSO (dimethyl sulfoxide). Mix the suspension by inverting the tube several times. Do not vortex.
Heat-shock the yeast by incubating the tube at 42°C for exactly 15 min. Swirl the tube occasionally to mix.
Chill the tubes on ice for 1 to 2 min.
Centrifuge the tubes at 2500 rpm for 4 min at RT.
Remove the supernatant, and wash the pellet in 0.5 ml sterile RT water by pipetting (do not vortex).
Centrifuge the tubes at 2500 rpm, then pipette all the supernatant off carefully.
Spread the yeast onto the appropriate media plate:
cDNA tubes (three): Resuspend each pellet in 700 μl sterile water. Spread 350 μl onto an SC-L-T plate (two plates/tube) to select for transformants.
No-DNA tube (one): Resuspend in 350 μl sterile H2O and plate onto SC-L-T. Spread the yeast by using sterile glass beads (Fisher Scientific International Inc., Pittsburgh, PA; 11-312A). For best results, shake the plates back and forth, not in a circle.
Place the yeast plates upside down at 30°C. Transformed yeast will grow in 2-4 d.
After colonies form, count the colonies on at least one plate to determine the transformation efficiency (colonies/μg DNA). The no-DNA control should have no colonies on the plate.
Yeast that grow on SC-L-T media contain the pGalcdk6 plasmid with the LEU2 marker and a cDNA library plasmid with the TRP1 selectable marker. The next step is to select yeast that may contain a protein-protein interaction between cdk6 and a protein from the library.
Selection for Interactors
The yeast are plated onto plates containing 3AT to select for yeast producing large amounts of histidine, which is an indicator of a protein-protein interaction. The number of cells inoculated onto selective plates determines the phenotypic differences between positive yeast and negative yeast. Thus, it is important to transfer a minimal number of yeast cells to the selective plates (see protocol below). The amount of time of incubation of the plate before transfer can also markedly influence the results.
After 48 h of incubation, transfer the colonies from the SC-L-T plates to SC-L-T-H + 20 mM 3AT selective plates by replica-plating using a sterile velvet. If no colonies are seen at 48 h, transfer colonies at 60 h. (Replica-plating stands and velvets can be purchased from Bel-Art Products, Pequannock, NJ.)
The inoculum on the selective plate is then diluted by replica-cleaning. This is the process of using sterile velvets to reduce the amount of yeast on the plate. In this procedure, a sterile velvet is put onto a replica-plating stand, and the SC-L-T-H 3AT plate is inverted onto the velvet with gentle pressure to remove some of the yeast from the plate.
In some cases, the plates should be replica-cleaned again after 24 h. Better transformation efficiencies result in heavier growth, and plates with such efficiencies will have to be replica-cleaned more diligently than plates with low transformation efficiencies.
Once potential “hits” (yeast that grow relatively more robustly on SC-L-T-H + 3AT) are identified, they are patched onto a new SC-L-T plate. This plate is commonly called the master plate (see Figure 3A). In addition to any potential hits, this plate is also patched with strong positive, weak positive, and negative control yeast. The strong positive contains the cDNAs of Fos in one plasmid and Jun in the second plasmid; the weak positive contains the cDNAs of Rb and E2F; and the negative control contains pGalcdk6 and an empty TRP1-containing vector (pPC86; available upon request). Depending on the number of hits isolated, students can share master plates because as many as 100 yeast colonies can be streaked onto one plate in a grid. This master plate is then replica-plated onto an SC-L-T + 20 mM 3AT plate and is used to perform an X-Gal assay as described below.
Gal-Lift Protocol or X-Gal Assay
Lift the yeast onto a MagnaGraph nylon membrane (Fisher Scientific, Catalog No. NJ0HY08550). After replica-plating the yeast onto an SC-L-T + 20 mM 3AT plate, place a circular nylon membrane on top of the yeast that have grown on the master plate. This procedure will transfer yeast from the master plate to the nylon membrane. Leave the membrane on the plate until the membrane has become moist. Carefully remove the membrane and place it on a YPD plate, yeast facing up, and incubate it at 30°C overnight.
The following day, prepare the following solution (wear gloves and goggles):
2.0 ml Z-Buffer
12 μl β-mercaptoethanol
30 μl (50 mg/ml) X-Gal (American Bioanalytical, Natick, MA; AB2400)
Immerse the membrane in liquid nitrogen by using tweezers to hold the membrane. Let the membrane thaw at RT.
Put the mixture from step 2 into the (upside-down) lid of a petri dish.
Put one Whatman #1 circular filter on top of this liquid and let it absorb the mixture.
On top of the filter paper, place the nitrocellulose membrane (yeast-side up) and cover it with the bottom of the petri dish, leaving the dish inverted with yeast facing up on the lid of the dish. Wrap the plate in aluminum foil and incubate it at 37°C for 18 to 48 h.
Check and record the results after 18-30 h of incubation. The strong positive control should be very blue, the weak positive slightly blue, and the negative control white.
Isolation of Plasmids
In this lab, yeast selected that contain potential interactors are lysed and DNA is isolated. To enrich for yeast containing the cDNA library plasmid (TRP1), yeast are first grown in SC-T, which allows the majority of the yeast to lose the pGalcdk6 plasmid.
Resuspend each positive yeast clone in 5 ml of SC-T media.
Incubate the culture for 18 to 24 h at 30°C at 250-300 rpm.
Centrifuge the culture at 2000 rpm for 5 min at RT.
Resuspend the cell pellet in 0.5 ml sorbitol reagent and transfer the mixture to a microcentrifuge tube.
Add 20 μl of 250 units/ml Zymolyase (Seikagaku America, Falmouth, MA; 120491) solution.
Incubate the tube for 60 min at 37°C.
Spin the tube in a microcentrifuge for 1 min at full speed (13,000 rpm). Discard the supernatant.
Resuspend the pellet in 500 μl of TE 50/20 solution.
Add 50 μl 10% SDS (Sodium dodecyl sulfate) and mix well.
Incubate the mixture for 30 min at 65°C.
Spin the tube for 5 min in a microcentrifuge at 13,000 rpm and save the supernatant to a fresh tube. The DNA is in the supernatant.
Add 700 μl isopropanol to the supernatant and mix. Incubate the mixture for 5 min at RT. The DNA can now be stored at 4°C, if necessary.
Centrifuge the tube for 10 s at 13,000 rpm (not more) in the microcentrifuge, decant the supernatant, then air-dry the DNA pellet for 10 min.
Resuspend the DNA pellet in 100 μl TE + RNAse (100 μg/ml). Incubate the tube for 30 min at 37°C.
Centrifuge the tube for 1 min at 13,000 rpm and transfer the supernatant to a fresh tube.
Following the manufacturer's instructions, use the GENECLEAN II kit (QBiogene, Inc., Carlsbad, CA; 1001-400) to purify the DNA for bacterial transformation.
Transform 20 μl of DNA into 100 μl competent bacteria.
At this point, bacterial transformation and alkaline lysis plasmid preparation can be done by standard protocols. Diagnostic restriction enzyme digestion can be used to differentiate the cDNA library plasmid from the pGalcdk6 plasmid. Once isolated, the cDNA can be sequenced to determine the protein interactor.
Yeast Media
Synthetic Complete (SC) Minus Tryptophan (SC-T) (1 l). In one 2-l flask, mix SC-L-T-H-U reagents:
1.7 g Yeast Nitrogen Base (DIFCO, DF0335-15) minus amino acids and without NH4SO4 | |||||
5.0 g NH4SO4 | |||||
1.0 g amino acid dropout mix (see below) | |||||
925 ml water | |||||
Adjust the pH to 5.9 with about 200 μl NaOH. | |||||
Autoclave 40 min. Let cool to at least 50°C and add the following before use:
50 ml 40% glucose | |||||
8 ml 20-mM uracil | |||||
8 ml 100-mM histidine | |||||
8 ml 100-mM leucine | |||||
Amino Acid Dropout Mix (-Uracil-His-Leu-Trp). Mix 5 grams each of the following: adenine sulfate, alanine, arginine, aspartic acid, asparagine, cysteine, glutamic acid, glutamine, glycine, isoleucine, lysine, methionine, phenylalanine, proline, serine, threonine, tyrosine, and valine. (All are available from Sigma Chemical Co., St. Louis, MO.)
Nutrient Solutions. Filter sterilize; do not autoclave.
20 mM uracil Store at RT. | |||||
100 mM histidine Store at 4°C. | |||||
100 mM leucine Store at RT. | |||||
40 mM tryptophan Store at 4°C. | |||||
SC-L-T. Follow above protocol, replacing 8 ml 100-mM leucine with sterile water.
YPD
10 g yeast extract (American Bioanalytical, AB01208) | |||||
20 g peptone (DIFCO, DF0118-17) | |||||
950 ml water | |||||
Autoclave 40 min. Cool to at least 50°C and add the following before use:
50 ml 40% glucose | |||||
For plates, add 0.1 g NaOH pellets and 20 g agar before autoclaving.
Yeast Plates. Add 20 g agar (DIFCO, DF0140-07) per liter of media. Add agar to media before autoclaving. Most protocols call for agar to be added to 450 ml water in a separate flask and autoclaved separately from the media (made up in a total volume of 475 ml). The contents of the two flasks are mixed after autoclaving. Media must be cooled to 50°C (a water bath set at 50°C works well), before glucose and liquid amino acids are added. Depending on the type of media made, volumes of water used are adjusted to reach a final volume (after addition of glucose and amino acids) of 1 l. It is widely believed that the quality of agar can influence transformation efficiency: DIFCO agar is widely considered the best.
Other Solutions
Z-Buffer (1 l). Use 16.1 g Na2HPO4 7H2O, 5.5 g NaH2PO4H2O, 0.75 g KCl, 0.25 g MgSO4 7H2O, and 2.7 ml β-mercaptoethanol. Adjust to pH 7.0 and bring to 1 l with water.
Sorbitol Reagent (100 ml). Use 18.2 g sorbitol (Sigma Chemical, S7547) and 20 ml 0.5-M EDTA. Adjust to pH 7.5 with NaOH and bring to 100 ml with water.
TE 50/20 (100 ml). Use 5 ml 1-M Tris pH 7.4, 4 ml 0.5-M EDTA, and add water to 100 ml.
Works Cited
Grossel, M., Wang, H., Gadea, B., Yeung, W., and Hinds, P. (1999). Cdk6 can shorten G1 phase dependent upon the N-terminal INK4 interaction domain. J. Biol. Chem. 274,29960 -29967.
Vidal, M., Brachmann, R.K., Fattaey, A., Harlow, E., and Boeke, J. (1996). Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions.Proc. Natl. Acad. Sci. USA 93,10321 -10326.
SAMPLE TWO-HYBRID PROJECT LAB REPORT
Introduction
The ultimate purpose of this experiment is to find out, through the use of the yeast two-hybrid system, information regarding the function of the protein cdk6. To learn more about this protein, an individual must identify, and then understand, the function of other proteins with which it associates. This is achieved by using the yeast two-hybrid system to express both the cdk6 protein and proteins encoded by a cDNA library. If and when cdk6 binds with a specific protein encoded by a cDNA library, the result will be transcription, visualized by the turning on of a reporter gene and the subsequent production of blue yeast. Transcription of a reporter gene depends on the binding of a functional transcription factor to DNA. The transcription factor prompts the RNA polymerase to begin transcription of the gene. Following the presence of blue yeast, indicative that a protein-protein interaction has occurred between cdk6 and the protein of interest (referred to as protein X), the identity of protein X will be determined through the process of first sequencing its cDNA gene by using the Sanger dideoxy method, and then scanning the BLAST database to find out whether the unknown gene was previously cloned. Finally, if the identity of protein X is known, its functions and role in the cell cycle can be researched and looked at in light of the function and role of cdk6, to ultimately provide more understanding of cdk6.
With this purpose in mind, it is important to realize that this experiment would not be possible without the use of a biological screen, which is beneficial when an individual is testing for specific protein-protein interactions. The idea behind a biological screen is that it allows proteins encoded by a cDNA library to be tested with a target protein in conjunction with a large number of other proteins rather than testing these possible hits individually. This experiment would not be possible if this method was not used because testing these interactions individually would take too long. This method provides the most effective method for finding the “needle” (interactions with cdk6) in the “haystack” (all possible proteins that are encoded by the cDNA library).
The complex events of the eukaryotic cell cycle are regulated by a select number of heterodimeric protein kinases. The regulatory subunits of these kinases are referred to as cyclins, and they have been determined to increase and decrease in phase with the cell cycle (Lodish et al., 2000). The catalytic subunits of cyclins are called cyclin-dependent kinases (cdks) because they have no kinase activity unless they are associated with a cyclin. Regulation of the eukaryotic cell cycle, specifically passage through the cell cycle, is controlled by G1-phase, S-phase, and mitotic cdk complexes. When cells are stimulated to replicate, G1-phase, cdk complexes are expressed, which prepare the cell for the S phase by activating transcription factors that cause expression of enzymes required for DNA synthesis and the genes encoding S-phase cdk complexes (Lodish et al., 2000). The S-phase cdk complexes phosphorylate regulatory sites in the proteins that form DNA pre-replication complexes, whereas mitotic cdk complexes induce chromosome condensation, breakdown of the nuclear envelope, assembly of the mitotic spindle apparatus, and alignment of condensed chromosomes at the metaphase plate, and eventually activate the anaphase-promoting complex (Lodish et al., 2000). Specifically, cdk6 has been the center of much research in part because of its ability to have a role in regulating G0/G1 and G1/S transitions. Cdk6 is important in regulation of the late G1 phase into the S phase because of its ability, in conjunction with cyclin D, to phosphorylate pRB. Phosphorylation of pRB protein is initiated by cdk4-cyclin D and cdk6-cyclin D in mid G1 phase. This initial phosphorylation leads to the generation of cdk2-cyclin E, which further accelerates the phosphorylation of pRB. Once phosphorylated, and thus inactivated, pRB is released from the transcription factor E2F. This transcription factor subsequently binds to nuclear DNA and turns on transcription of S-phase genes that allow replication. Arbogast et al. (1999) investigated the possibility that a loss of activity of cdk6 was implicated in the age-related dysfunction of the cell cycle in its initial phase. Following the conclusion of their research, it was determined that the low cdk6 activity in T lymphocytes from older persons was associated with a defective phosphorylation of the endogenous Rb protein and an increased sequestration of the E2F-1 transcription factor. It is possible that this results in early cell cycle arrest.
Also important in the two-hybrid screen are the specific proteins, which are encoded by a library of genes known as a cDNA library. cDNA stands for complementary DNA, which are DNA copies of mRNA. Throughout, cDNA clones were dealt with, which were made prior to beginning the experiment. cDNA clones lack the noncoding introns present in genomic DNA clones. Consequently, the amino acid sequence of a protein can be determined directly from the nucleotide sequence of its corresponding cDNA. Crucial to the production of a cDNA library is the enzyme reverse transcriptase. After the mRNA is isolated from the tissue of interest, reverse transcriptase is then used to synthesize a strand of DNA complementary to each mRNA molecule. The enzyme works by polymerizing deoxynucleoside triphosphates into a cDNA strand by using the RNA molecule as a template. It is important to note that after going through the process of transcription and translation, one cDNA encodes only one protein. The cDNA library is one of the plasmids present within the yeast colony; the other is Galcdk6. Ideally, one of the proteins encoded by the cDNA library specifically interacts with cdk6. The likelihood of this interaction is purely by chance, but it is helped by the fact that the library was made from RNA from a human brain and thus is likely to contain most proteins. Specifically, the human genome spans 40,000-50,000 genes that code for proteins, and of these 40,000-50,000, between 5000 and 10,000 are expressed in the human brain.
The two-hybrid system that is used is based on the system developed by Vidal et al. (1996) in Ed Harlow's lab. In essence, the two-hybrid system uses transcription as a tool to determine whether a protein-protein interaction occurs between cdk6 and an unknown protein. Under normal cellular conditions, transcription occurs when transcription factors bind DNA, and the transcriptional-activation domain (TAD) causes polymerase to activate gene expression at the transcription initiation site. A transcription factor is made up of two parts: a DNA-binding domain (DBD) and a TAD. Although each factor can act alone, both are necessary to activate a gene. This principle is used in this assay, in which the DBD and the TAD are separated and are brought back together only if cdk6, bound to the DBD, binds to protein X, which is bound to the TAD. If they are brought back together, and the original transcription factor is formed, transcription will occur. Prior to the beginning of the experiment, preliminary steps were done. These steps included cloning the cDNA of cdk6 into the correct plasmid vectors, which allowed them to be expressed in the yeast system. In addition, the cdk6 was made to have a Gal4 DBD. The fusion proteins were tested to ensure that they made the expected-size proteins and could activate the reporter gene only when they were bound to another protein.
In this experiment, the yeast strain used was HW18 + pGalcdk6. The two hybrids were produced by two vectors, one encoding for cdk6 bound to the Gal4 DBD. Here, the cdk6 protein acts as the bait in the system. This plasmid contained a promoter, the DBD, cdk6, a nuclear localization signal, a leucine marker, an ampicillin site, and both a yeast and a bacterial origin of the replication site. The second plasmid codes for protein X, which acts as the prey in the system. Protein X is coded for by the cDNA present in the plasmid, bound to a TAD. The cDNA plasmid was similar except for the cDNA and the TAD fragment in place of the cdk6 and the DBD and except for a tryptophan marker instead of leucine.
The first step in successfully screening the cDNA library is to transform the library into the yeast cells. Because the Gal-cdk6 plasmid was already present in the nucleus of the yeast cell, it was our responsibility to see that the cDNA library plasmid was correctly transformed into the cell. To work with conditions that allowed for the most efficient transformation, we had to capture the yeast at the logarithmic stage of growth, which was determined to be at an optical density 600 nm as close to 0.3 as possible. The process of transformation was carried out by using sterile techniques, and selection was used to pick yeast that contained both plasmids.
A very essential aspect of this system is the proper use of selection. The notion behind this process is allowing for the growth of the yeast that contain plasmids with the specific markers, while getting rid of the yeast that did not transform. There were two different types of selections used for working with the yeast and the Escherichia coli bacteria. Plating the bacteria on a medium containing AMP following bacterial transformation allowed the bacteria that did not transform to die. Conversely, during selection for yeast, a medium was used that lacked specific amino acids necessary for the cell to survive. Thus, the only way for the yeast to survive on the medium is to produce the amino acid. This is possible because the specific amino acids, leucine and tryptophan, are present within the cDNA and Galcdk6 plasmids, respectively. Plating the yeast onto synthetic complete (SC) media lacking tryptophan and leucine allows selection to occur for the yeast that have both plasmids. The selection process was utilized prior to the start of the experiment by plating the yeast onto SC media without leucine so that cells with Galcdk6 plasmids would grow on the plate.
The selection process is used again during replica-plating onto SC-L-T-H + 10 mM 3AT (3-amino-triazole) as a method for testing reporter gene transcription. This selects for yeast that have the desired protein-protein interaction between cdk6 and the unknown protein X because yeast with this interaction produce histidine. The HIS3 gene is located on the genome of the yeast and encodes imidazole glycerol phospate dehydratase, which is an enzyme involved in histidine biosynthesis. This HIS gene is referred to as a reporter gene because its presence indicates that the interaction occurred. By growing on a medium that lacks histidine, only the yeast that are producing histidine, and contain the interaction, remain. The presence of the 3AT is crucial to the selection process because of its function as an inhibitor for the enzyme imidazole glycerol phosphate dehydratase. Consequently, the presence of 3AT serves as a further selection for the yeast with the desired interaction. Only the yeast that are producing a lot of histidine, because of the “strong” interaction, can overcome this inhibitor and survive on the medium. When the concentration of the 3AT was increased, and the potential positives gridded, this selection allowed the yeast that were actually producing histidine to be pinpointed. The amino acid-based selection was used for a final time when positives were resuspended in a medium of SC-T. This enhances for the cDNA plasmid containing tryptophan by increasing the likelihood that the yeast will keep the plasmid and release the cdk6 plasmid.
As stated earlier, this two-hybrid system is centered around the processes of transcription and translation. Although transcription is more important in the overall outcome, translation is necessary for the production of the transcription factors. Transcription and subsequent translation of the two plasmid genes produces both the DBD-Galcdk6 and the TAD-protein X complexes. The presence of a nuclear location signal brings the resulting proteins back into the nucleus of the yeast cell from the cytoplasm where translation occurred. If the desired protein-protein interaction occurs within the nucleus, the resulting transcription factor binds to the promoter region of the reporter gene and enhances the transcription of either lacZ or HIS genes, located on the yeast chromosome. Specifically, the lacz operon produces mRNA that codes for an enzymeβ -galactosidase, which catalyzes the hydrolysis of lactose into galactose and glucose. However, in this experiment, this enzyme reacts with X-Gal in an enzyme substrate reaction to form a blue product that is visible in an assay. In the presence of an alternate substrate, 5-bromo-4-chloro-3-indol-β-d-galactopyranoside (X-Gal), the product of the enzymatic reaction is 5-bromo-4-chloro-indoxyl, which dimerizes to form a blue product. In essence, β-galactosidase cleaves the substrate, X-Gal, which thus produces the blue color.
The presence of the blue color indicates that the clones of yeast have given a positive reporter indication for interaction with cdk6. Thus, it is necessary pick out the cDNA plasmid so that the cDNA can be isolated and sequenced. The process of isolation was by means of a smash-and-grab procedure. This was then followed by bacterial transformation, which is a means of separating the two plasmids. Restriction analysis of these plasmids, followed by gel electrophoresis, allowed for the differentiation between cDNA and Galcdk6 plasmids. The cDNA plasmids were then sequenced by using the Sanger method, and the resulting information was used in a BLAST search of protein identities. This knowledge can then be used to make conclusions about the cellular functions of cdk6.
Materials and Methods
Yeast Transformation. The morning of lab, HW18 yeast was resuspended in 5 ml sterile water and vortexed to disperse any clumps. Then 50 ml of YPD + Glucose culture was inoculated with yeast to OD600 = 0.130 Å. This culture was grown until OD600 = 0.30 to 0.34. Fifty milliliters of yeast at OD600 = 0.207 was poured into a 50-ml conical tube. This was spun in the tabletop centrifuge at 1800 rpm at 20°C for 4 min. The media (supernatant) was then poured off into a liquid waste container, and the yeast pellet was resuspended in 20 ml of sterile room temperature water. The resuspension was performed using appropriate sterile techniques. After resuspension, the tube containing the yeast pellet was spun at 1800 rpm at 20°C for 4 min. The supernatant was poured off, and the yeast was resuspended in 4.5 ml of freshly prepared TE/LiAc solution. A total of 40 ml was prepared from the stock solutions and contained 4 ml 1-M LiAc, 0.4 ml 1-M Tris, 0.08 ml 0.5-M EDTA (ethylenediamine tetraacetic acid), and 35.52 ml sterile water. The yeast and this solution were spun at 1800 rpm at 20°C for 4 min. The supernatant was poured off, and the yeast was then resuspended in 250 μl of fresh TE/LiAc solution. Four tubes were prepared using the following: 1 μg of 1 μg/μl of cDNA library plasmid DNA, 5μ l of 10-mg/ml denatured single-stranded salmon testes DNA (ssDNA); 100μ l of the yeast prepared above was added and vortexed, and then 600 μl of freshly made PEG (polyethylene-glycol) was added, and the mixture was vortexed again. The PEG solution consisted of 40% PEG, 10 mM Tris, 1 mM EDTA, and 0.1 M LiAc. All four tubes were then incubated at 30°C for 30 min. Seventy microliters of sterile DMSO (dimethyl sulfoxide) was added to each tube, and the tubes were inverted. The cells were then heat-shocked by incubating them at 42°C for exactly 15 min. During the 15 min, the tubes were swirled four times. After the 15 min, the tubes were immediately placed on ice for 2 min. Next, the tubes were spun at 2500 rpm for 4 min at room temperature. The supernatant was removed, the pellet was washed in 0.5 ml sterile water by pipetting up and down, and the tubes were then spun again at 2500 rpm for 4 min. The supernatant was pipetted off carefully. For the cDNA library tubes, each pellet was resuspended in 700 μl of water, and each tube was plated onto two plates of SC-L-T using 350 μl/plate for a total of eight plates. Glass beads were used to spread and allow for even growth by shaking the plate back and forth. The yeast plates were then labeled with name and date, taped together, and placed into the 30°C incubator for 3 d.
The eight SC-L-T plates were removed from the incubator, and seven of the plates were replica-plated and replica-cleaned onto seven SC-L-T-H + 10 mM 3AT plates. These plates were grown at 30°C and were replica-cleaned after 1 d. The eighth plate was placed in a 4°C refrigerator to determine transformation efficiency. The next day, because of overgrowth, the seven plates were replica-plated onto a new set of seven SC-L-T-H + 10 mM 3AT plates.
Gridding Positive Yeast. Forty-eight hours later, after being grown at 30°C, the transformation efficiency was determined by calculating the number of colonies/μg cDNA library. A master plate was made by gridding potential positive colonies from the seven SC-L-T-H + 10 mM 3AT plates onto SC-L-T plates, and the second set of seven SC-L-T-H plates were replica-cleaned again. The plates were incubated at 30°C. Twenty-four hours later, another set of potential positives from the second replica-cleaning were gridded onto SC-L-T plates to create a second master plate. The seven SC-L-T-H + 10 mM 3AT plates were discarded. The two master plates were then replica-plated onto SC-L-T + 20 mM 3AT plates 24 h later. The yeast patches were also lifted onto MagnaGraph nylon membrane (Fisher Scientific). This lifting was performed by first putting a membrane down on the master plate until it was moist; then, once it was removed, the membrane was placed on a YPD plate with the yeast side up and incubated at 30°C overnight. The master plates were stored at 4°C. As a result of difficulties performing the Gal lift by the next day, the plates were removed from the incubator and placed in the 4°C refrigerator 24 h after being placed in the 30°C incubator.
Gal-Lift Protocol. The following was performed for both membranes. On 11/1/01, the Gal lift was performed by first preparing a solution consisting of 2.0 ml Z-Buffer, 12 μl β-mercaptoethanol, and 150 μl X-Gal (20 mg/ml). Tweezers were used to lift the membrane from the YPD plates, immerse it into liquid nitrogen, and subsequently place it on a paper towel to let it dry/thaw. The mixture described above was put into the lid (upside down) of a petri dish. A Whitman circular filter was placed on top of this liquid, which allowed it to become evenly wet. Once dry, the nitrocellulose membrane was added, yeast-side up, and then covered with the bottom of the petri dish. The plate was wrapped in aluminum foil to prevent it from drying and was placed in the incubator at 37°C for 24 h. The X-Gal was returned to the freezer, and the liquid nitrogen secured. The plates were removed from the incubator 24 h later, and the results checked.
Plasmid DNA Extraction from Yeast Transformants. On 11/7/01, two potential positives were selected from the SC-L-T master plate with a sterilized toothpick, and each was resuspended in 5 ml of SC-T media in a snap-cap tube. A different toothpick was used to scrape each of the two yeast clones and then dropped into the 5 ml of culture. The snap-cap tube was placed in the shaking incubator at 30°C at 250 rpm for 24 h.
After the 24 h, the snap-cap tube was removed from the shaking incubator, and the 5 ml of yeast was spun down in the tabletop centrifuge for 5 min at 2000 rpm. The supernatant was discarded, and the cell pellet was resuspended in 500 μl of sorbitol reagent and transferred into microcentrifuge tubes. Twenty microliters of Zymolyase solution was added to each tube, and the microcentrifuge tubes were placed in the 37°C incubator for 60 min. Next, the microcentrifuge tubes were spun at full speed for 1 min and the supernatant was discarded. The pellet was then resuspended in 500 μl of TE 50/20 solution, 50 μl of 10% SDS was added, and the solution was mixed and then placed in a 65°C incubator for 30 min. The microcentrifuge tubes were removed from the incubator after the 30 min, 200 μl 5-M Kac was added to each solution, and then the tubes were placed on ice for 60 min. The tubes were removed from the ice and spun at full speed for 5 min. The supernatant was then pipetted into a fresh tube. Seven hundred microliters of isopropanol was added to each supernatant and mixed; then, the contents were incubated at room temperature for 5 min.
The microcentrifuge tubes were then spun for exactly 10 s at full speed, the supernatant was poured off, and the pellet was air-dried for 10 min. To help with the drying process, a Kimwipe (Kimberly-Clark Corp.) was used to absorb extra moisture. The DNA pellet was resuspended in 100 μl of TE RNAse at a concentration of 100 μg/ml and then incubated for 30 + min at 37°C. The tubes were then spun for 1 min, and the supernatant was transferred to new, labeled tubes. After spinning, 300 μl NaI solution and then 10 μl of Glassmilk (BIO 101) were added to each tube. The DNA was then allowed to bind to the Glassmilk for 5 min at room temperature while occasionally being vortexed. Next, the tubes were spun at full speed for 2 min, then the Glassmilk/bound DNA pellet was washed three times with 500 μl of New Wash (BIO 101). After the third wash, the Glassmilk pellets were redissolved in 50 μl of TE. The tubes were spun for 1 min, and the supernatant containing the plasmid DNA was saved into a new, labeled tube. The process of transforming 10 μl into 50 μl of competent bacteria was performed and is described below.
Transformation of Competent E. coli with Plasmid DNA. A tube of competent cells was obtained from an ice bucket. The transformations were performed by mixing 10 μl of plasmid DNA samples with 50 μl of competent E. coli cells. The cells were then incubated on ice for 30 min. The cells were then heat-shocked for exactly 90 s by placing the microcentrifuge tubes in a 42°C water bath. After the 90 s, the cells were immediately placed on ice. Eight hundred microliters of LB (Luria Bertani Broth) + Glucose was added to each tube, and the cells were then placed in a shaking incubator at 37°C for 45 min. The cells were next spun at a setting of 7 for 1 min. Then, all but 200 μl of supernatant was removed, and the pellet was resuspended in the remaining LB by pipetting up and down. One hundred eighty microliters of this cell slurry was then pipetted onto an LB/AMP (Ampicillin), labeled plate. Twenty microliters of transformed cells was plated on one LB plate without AMP. With the use of proper sterile techniques, the slurry was evenly spread on the plate with the help of a glass wand and a spinning device. The plates were then placed in the 37°C incubator, upside down, for 24 h. The next day, the plates were removed from the 37°C incubator and placed into the 4°C refrigerator.
Preparation of DNA to Sequence. A bacteria colony was picked with a toothpick and dropped into the 5 ml of BHI (brain-heart infusion)/AMP. The inoculated cultures were then placed into the 37°C shaking incubator. This procedure was performed for us and repeated a number of times for individual tubes.
An E. coli tube containing DL1 sample was obtained, and the procedure was followed for the Qiagen-Prep DNA. A flask of 1% agarose gel + EtBr (Ethidium Bromide) was obtained. This was prepared by mixing 1 g LE agarose with 100 ml of 1X TBE (Tris Borate EDTA) and was then heated in a microwave for 3 min with a Kimwipe plug to prevent moisture loss. When the solution cooled, 3 μl of 10 mg/ml EtBr was added. Because the gel had solidified, it was reheated in the microwave until it turned to liquid. When the gel cooled, it was poured into the electrophoresis box, which contained a plastic stopper and an eight-piece comb. After the gel had solidified, 100 ml 1X TBE, which consisted of 20 ml 5X TBE + 80 ml distilled water, was poured on top and the comb was removed.
A 20-μl total volume enzyme digest was prepared by following six steps: 1) Nine microliters of distilled water, 5 μl plasmid DNA, 2 μl NEB Buffer 3 (New England BioLabs Inc., Beverly, MA), 2 μl 10X BSA (Bovine Serum Albumin), 1 μl Not I, and 1 μl Mlu I were added to a microcentrifuge tube. 2) The mixture was then incubated at 37°C for 60 min in a heating block. 3) Four microliters of 6X loading dye was added to the reaction mixtures, and a prepared sample of 500 μg/ml 1-kb DNA standard was obtained. 4) Twenty microliters of the samples was loaded into the gel wells, the lid was closed, electrodes were attached, and the gel was run at 125 V until the bromophenol blue was halfway down the gel. 5) The gel was then removed and placed on the UV box for visualization. 6) A Polaroid picture was taken.
Results
Transformation. Yeast strain HW18 + pGalcdk6 was transformed by using plasmid that contained the cDNA library and a tryptophan marker. The cDNA library was 5000-10,000 genes from the human brain. As a way to enhance for transformation of the cDNA plasmid into the yeast containing the Galcdk6, the yeast containing the Galcdk6 plasmid was grown on a medium of SC-L. The yeast containing the Galcdk6 plasmid was inoculated in a YPD+ Glucose culture, which grew to OD600 = 0.307 in approximately 8 h. Specifically, the yeast was transformed when the optical density reached mid-log phase. The growth of the yeast follows a logarithmic scale, beginning at the lag phase with an OD600 = 0.130 Å, and then followed by logarithmic growth until it eventually levels off. The objective is to transform the yeast at an optical density as close to 0.3 as possible, determined to be the mid-log. This allows for conditions suitable for the most effective transformation possible. Making the yeast competent to take up the cDNA required the addition of TE/LiAc, a solution consisting of LiAc, Tris-HCl, and EDTA. The actual transformation process required the addition of cDNA library, TE/LiAc, (ssDNA) salmon sperm DNA, and PEG solution. The PEG, which prevented the cells from freezing, was added last because if added in the beginning it would cause the DNA to precipitate. The presence of the ssDNA was important because this DNA is easy to isolate, will separate into single strands, and, most important, acts as a carrier that helps the yeast take up the DNA. DMSO was also added and allowed the membrane of the yeast cells to become permeable. Heat-shock, which provided optimum conditions for plasmid uptake, followed by recovery on ice, finished the transformation process. As a way to select for the yeast that transformed, they were plated on the medium SC-L-T. As a means of comparison to determine whether the proper experimental conditions existed, Sarah Morgan performed a negative control, which contained the same contents as did the cells that were to be transformed, except the cDNA library plasmid.
Selection. The plating onto SC-L-T was followed by an additional selection, which consisted of plating all seven dishes onto SC-L-T-H + 10 mM 3AT. Because of the high transformation efficiency, the seven original plates were replica-plated and replica-cleaned onto a new set of seven SC-L-T-H + %10 mM 3AT plates, depicted in Figure A1 as the right-hand dish. To determine the actual transformation efficiency, we counted the number of colonies on the SC-L-T plate and expressed them as the number of colonies per microgram of library DNA (Table A1). Colonies that strongly grew on the media containing the 3AT are transcribing the reporter gene, and thus having the protein interaction between protein X and the cdk6 protein. Cells with this interaction survived in this test for the HIS3 reporter gene because they overcame the inhibitor and produced His.
| One-eighth of the plate consisted of 254 yeast colonies. |
| 254 × 8 = 2032 yeast colonies on the plate |
| 2032 × 2 = 4064 transformed yeast/μg cDNA |
| 4064 × 4 = 16,256 colonies/μg of library DNA |
Figure A1. (Top) Master plate. (Bottom right) Gal lift done on nitrocellulose. (Bottom left) Replica-plate of master onto SC-L-T-H + 20 mM 3AT.
A master plate was prepared by streaking potential positive colonies from the two sets of seven SC-L-T-H 10 mM 3AT onto their respective master plate containing+ SC-L-T media. Although difficult to visualize by looking at Figure A1, these potential positives appeared stronger by standing out in terms of larger size from the other colonies. In addition to the potential positives, a control was used by streaking a positive, weak control, and a negative (from right to left) on the top row of the master plate as a means of determining whether the growth conditions were accurate. As evident by looking at the top plate in Figure A1, the positive control grew the best and the negative appeared the weakest. The grid of the potential positives streaked on the master plate is used to test the reporter genes. The potential positives that were streaked on the master plate grew on media containing 3AT, and theoretically should have taken up a cDNA that makes the protein that binds to cdk6. As a way to perform the actual test determining the presence of the second reporter gene, lacZ, the two master plates were lifted onto individual nitrocellulose membranes and then placed on a YPD plate. The nitrocellulose is a membrane permeable to nutrients and acts as a transporter for the yeast. The nitrocellulose was immersed in liquid nitrogen designed to pop open the yeast and expose the enzyme to undergo an enzyme substrate reaction. The lacZ gene, produced by the operon when the protein-protein interaction occurs, encodes the β-galactosidase enzyme that interacts with the X-Gal substrate to produce a blue color. In addition to the X-Gal, the presence of the Z-Buffer and β-mercaptoethanol allowed the enzyme substrate reaction to take place. The nitrocellulose located on the bottom right of Figure A1 illustrates another intent of the control in the sense that the positive turned blue, the negative did not, and the weak positive was somewhere between. This finding indicates that the X-Gal reaction conditions were proper. The cdk6-protein X interaction of interest can be visualized by looking for blue-colored yeast on the nitrocellulose paper. Figure A1 illustrates the absence of blue color. In terms of total class results, 189,000 transformed yeast were screened, which led to five successful cdk6-protein X interactions. Also, a selection was performed from the master plate by replica-plating the master onto a medium containing SC-L-T-H + 20 mM 3AT to further select for those producing histidine.
Plasmid Recovery. Although no positive reporter indication was given on the nitrocellulose, two possible positives were picked from the SC-L-T plate on the basis of growth characteristics and were resuspended in SC-T media. As explained before, the SC-T media enrich for yeast containing the cDNA plasmid by providing for a better than 50% chance that the cDNA plasmid, rather than the cdk6 plasmid, will be kept by the yeast. A smash-and-grab procedure was carried out to separate the DNA plasmid from the yeast chromosome as well as from the other plasmid that contains Galcdk6. To help with this, sorbitol reagent was used, which is a salt that creates a hypotonic environment for the yeast. Similar to plants, yeast contain a cell wall-like exterior, which needs to be “chewed up” in order to“ gain access” to the plasmids. This was accomplished with the addition of Zymolyase solution. Also added was SDS (Sodium dodecyl Sulfate), which was responsible for denaturing the yeast proteins, while Kac functions similarly to NaOH and denatures the chromosomal and plasmid DNA. Denaturing is necessary because it makes precipitation possible using isopropanol.
Exposure of the plasmids was followed by transformation into bacteria cells. Transforming into bacteria is a process of selection in and of itself because E. coli is capable of taking up only one plasmid. Transforming into bacteria is also beneficial because bacteria cells amplify, which in effect amplifies the plasmids inside. The bacteria cells used were made competent by CaCl2 prior to beginning the procedure. However, before introduction of the competent cells, the RNA was eliminated with RNAse, and the DNA left in solution was washed away by means of binding to Glassmilk. The presence of NaI changed the pH to allow the DNA to bind to the Glassmilk. The addition of the New Wash precipitated the DNA, and the subsequent addition of TE cleaved the plasmid DNA from the Glassmilk. Consequently, the resulting solution contained the isolated plasmids. As a means of selection, these isolated plasmids were then introduced to the competent bacteria cells and then heat-shocked to allow the actual uptake of either plasmid into the bacteria cell. Although selection is an important step after transformation, no AMP was used when the cells were first plated on LB and glucose because those conditions would be too strong for the newly transformed and still fragile cells. Following a period of recovery, the bacteria cells were then plated on an LB/AMP medium to allow for selection of the bacteria cell that did not take up the plasmid from those that did. At this stage, transformants survived and cells without a plasmid died. The removal of all but 200 μl, followed by resuspension of the pellet in the remaining LB, allowed for a more concentrated solution by decreasing the solvent and increasing the solute. Also, a plate was made that did not contain AMP to assess for growth efficiency, and all plates were placed in a 37°C incubator, upside down to prevent condensation.
Colonies did appear on the plate, but the focus was now shifted onto positive hits from the class. The remainder of the experiment was performed on another student's hit. The following procedure was performed on sample DL1. Colonies from the LB + AMP plate were picked and grown in BHI + AMP media for us. The DNA was prepared by method of Qiagen, which was then followed by an enzyme digest to determine whether the library plasmid contained an insert. A restriction digest was performed by using Not I and Mlu I restriction endonucleases to determine whether the plasmids were cDNA or Galcdk6 (Figure A2). If the cDNA library plasmid was present, the result would be two bands; the smaller of the two would represent the cDNA insert because both restriction sites for Not I and Mlu I exist (Figure A3). However, the Galcdk6 plasmid would linearize because only the Not I site is present (Figure A3). In addition, NEB Buffer 3 (New needs to be added for the Mlu I, while Not I requires both NEB Buffer 3 and BSA. The compatibility of the two restriction endonucleases and the need for the buffers were determined by referencing the NEB manual. After the enzyme digest was performed, the DNA was run on a gel so that we could determine the size of the bands and thus make subsequent conclusions as to which plasmid was present and whether the cDNA insert was present. This double digest should result in a linearized band representative of the cdk6 plasmid because of the presence of only one endonuclease, Not I. The cDNA plasmid should appear as three bands on the gel after the double digest because of the presence of both endonucleases. The band produced by cutting from Mlu I to Mlu I is the smallest of the bands and too small to be visible on the gel. Mlu I to Not I generates the cDNA insert as the second smallest band, whereas cutting from Not I to Mlu I generates the largest band, which is thus highest up on the gel. Following the restriction digest, the samples were run on a 1% agarose gel with EtBr, and the subsequent results were analyzed. Figure A2 shows a Polaroid picture of the gel. Lane 1 is located on the left-hand side, with the other lanes following in numerical order. Lane 1 contains the 1-kb DNA ladder, while lane 2 contains MB4; lane 3, JL2; lane 4, DLI; lane 5, JL2 (A); and lane 6, JL2 (B). Lanes 7 and 8 are empty. As illustrated by Figure A2, no DNA was evident in lanes 2, 3, and 5, whereas a single band around 9 kb appeared in lane 6. Lane 4 also showed a band around 8 kb (cDNA plasmid), and a faint, thin band also appeared around 1.2 kb, probably representative of the cDNA insert. This top 8-kb band is close to the expected 7093-kb value of the cDNA plasmid. The single, approximately 8-kb band present in lane 6 is theoretically indicative of the cdk6 plasmid. This molecular weight value, based on reference to the 1-kb DNA ladder, is close to the expected value of the cdk6 plasmid, which is 8.3 kb. Furthermore, only one band is evident, which indicates a linearized plasmid. Visually, Figure A2 suggests that lane 6 contains the cdk6 plasmid and lane 4 the cDNA plasmid, and thus the DLI insert. However, although lane 6 matches the criteria for the cdk6 plasmid, it is probably representative of a cDNA plasmid. The reason for this is because the two top bands in lanes 4 and 6 match in terms of molecular weight. In addition, it was previously established that the 1.2-kb band in lane 4 was representative of the cDNA insert. Thus, although only one band is evident in lane 6 (Figure A2), one can infer that the visible band is the cDNA plasmid. It is possible that the insert was present but that there was not enough DNA for it to be visible. It is also possible that the insert from JL2 was missed because the insert was smaller than the 0.5-kb marker. Although this reason is not as likely as the first, it is nevertheless a possibility because the molecular weight of that insert would still not be too far from the library average of 2.1 kb.
Figure A2. One percent agarose gel. Lanes are assigned from left to right in numerical order. Lane 1 contains the 1-kb DNA ladder; lane 2, MB4; lane 3, JL2; lane 4, DL1; lane 5, JL2 (A); and lane 6, JL2 (B). Lanes 7 and 8 are empty.
Sequencing. On the basis of the findings from Figure A2, it appears that there is a cDNA insert present in DLI. As a way to determine the identity of the insert, the remaining solution left over from loading the gel will be used in sequencing. In addition to the insert, dNTPs (deoxynucleoside triphosphates), ddNTPs (dideoxynucleoside triphosphates), primers, and DNA polymerase were added to allow for the proper functioning of the Sanger dideoxy method of sequencing. A printout was generated that provided the sequence of the insert.
Works Cited
Arbogast, A., Boutet, S., Phelouzat, M., Plastre, O., Quadri, R., and Proust, J.J. (1999). Failure of T lymphocytes from elderly humans to enter the cell cycle is associated with low cdk6 activity and impaired phosphorylation of Rb protein. Cell. Immunol. 197,46 -54.
Grossel, M. (2001). Molecular Biology Experiments and Research, 3rd ed. New London, CT: Connecticut College,18 -40.
Lodish, H., Berk, A., Zipursky, S.L., and Matsudaira, P. (2000). Molecular Cell Biology. New York: Freeman,219 -228,496 -498.
Vidal, M., Brachmann, R.K., Fattaey, A., Harlow, E., and Boeke, J. (1996). Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions.Proc. Natl. Acad. Sci. USA 93,10321 -10326.
SAMPLE TWO-HYBRID PROJECT POSTERS
Figure A3. Plasmid maps of (A) Galcdk6 and (B) cDNA library plasmids.


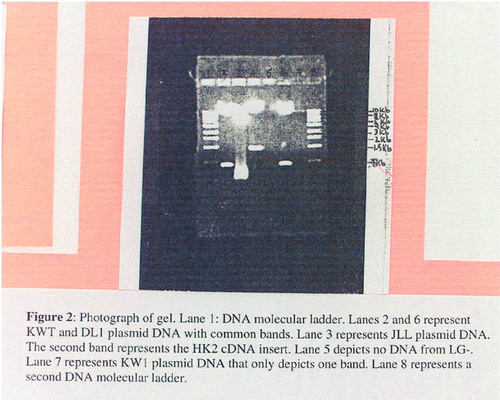
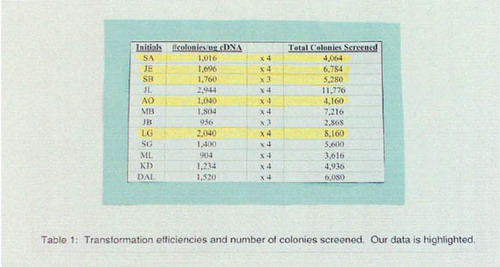
SAMPLE TWO-HYBRID PROJECT FINAL PAPER
KIF2 is a motor protein that transports organelles and other various cellular components throughout the cell along microtubules, using energy from hydrolyzed ATP (adenosine triphosphate). The heavy chain region of this heterodymeric protein contains a microtubule binding site/motor domain along with a site for ATP hydrolysis, whose activity is said to be regulated by the light chain (Verhey and Rapoport, 2001).1 This head-to-tail self-inhibition is relieved upon the binding of a kinase, which causes a conformational change allowing the light chain to move away from the ATP-hydrolysis site and activation of KIF2 (Verhey and Rapoport, 2001). This activation results in KIF2 motility toward the + end of microtubules, specifically associated with “anterogradely transported membranous organelles in nerve axons” (Hirokawa, 1998; Morfini et al., 1997).2,3
1Verhey, K.J., and Rapoport, T.A. (2001). Trends Biochem. Sci. 26(9), 545-549.
2Hirokawa, N. (1998, January 23). Science 279, 519-526.
3Morfini, G., Quiroga, S., Rosa, A., Kosik, K., and Caceres, A. (1997, August 11). J. Cell Biol. 138(3), 657-669.
Takemura (1996)4 and Hirokawa (1998) concur that KIF2 is involved in axonal extension or outgrowth of developing neurons, which explains its ubiquitous expression in juvenile brain but not in adult neurons. KIF2 has been found to transport vesicles containing the β-gc portion of a growth cone membrane receptor for IGF-1 (Insulin-like Growth Factor 1), a growth factor that is essential in nerve cell development (Morfini et al., 1997). Experimentation has proven that KIF2 suppression results in a buildup of β-gc in the cell body of the neuron, which results in the inhibition of neurite extension (Morfini et al., 1997). Morfini et al. have proposed that the “mechanism by which KIF2 participated in neurite extension... relates with the nature and final destination of the cargo that it transports.”
4Takemura, R., Nakata, T., Okada, Y., Yamazaki, H., Zhang, Z., and Hirokawa, N. (1996). J. Neurosci. 16(1), 31-35.
The known role of cdk6 during the G1 phase of the cell cycle is to bind cyclin D, which together phosphorylate a Rb/E2F complex, which allows the release of E2F. E2F is a transcription factor that turns on the transcription of S-phase genes so that DNA can be synthesized and the cell cycle (cell division) can continue. However, little is known about the function of cdk6 after the G1 phase of the cell cycle and in postmitotic cells. The initial connection between cdk6 and KIF2 is that KIF2 mobility is initiated by the binding of a kinase, such as cyclin-dependent kinase 6 (cdk6). Cdk6 binding allows KIF2 to travel down the axon, similar to traveling in the + end direction of a microtubule, to deliver necessary growth factor receptors to the growth cone of the neurons. Furthermore, inhibition of cdk6 is known to induce mitotic arrest, which allows for terminal mitosis and neural differentiation (Ferguson et al., 2000).5 Thus, it is hypothesized that once nerve cells have undergone terminal mitosis, cdk6 has finished its job as a regulator in the G1 phase of the cell cycle. To prevent further division of these differentiated cells, KIF2 competes with any cyclin D that has not undergone ubiquitination to bind with and inhibit cdk6. Besides mitotic suppression, a second benefit of cdk6-KIF2 binding is that the activation of KIF2 allows anterograde transport of growth factor receptors to the growth cones at the terminal end of the axon, which promotes neurite outgrowth (Morfini et al., 1997).
5Ferguson, K.L., Callaghan, S., O'Hare, M., Park, D., and Slack, R. (2000, October). J. Biol. Chem. 275(43), 33593-33600.
If this hypothesis is correct, an increased amount of cdk6 and KIF2 should increase the distribution of β-gc in the axon terminal growth cones. Neurons transfected with cdk6 expression vectors (Grossel et al., 1999)6 will up-regulate cdk6 expression. This can be proven by immunoblot analysis (Morfini et al., 1997) of cdk6 from these cells along with cdk6 from cells transfected with “empty” vectors (for control) with antibodies against cdk6. The amount of KIF2 expression after NGF up-regulation can be examined by immunoblot using antibodies against KIF2 (Morfini et al., 1997) and compared with control cells not treated with NGF. If the subcellular distribution of β-gc in the growth cones of NGF-treated cdk6 vector-transfected cells is higher than that in NGF-treated empty vector-transfected cells or in non-NGF-treated cdk6 vector-transfected cells, then the hypothesis is proven correct. The subcellular distribution ofβ -gc can be investigated by means of double immunofluorescence micrographs showing the distribution of β-gc (Morfini et al., 1997).
6Grossel, M.J., Baker, G., and Hinds, P. (1999, October). J. Biol. Chem. 274(42), 29960-29967.
FOOTNOTES
Monitoring Editor: Elizabeth Vallen
ACKNOWLEDGMENTS
We thank Stephen Loomis for critical reading of the manuscript. We also thank Andrew Warren for excellent technical assistance. This material is based on work supported by the National Science Foundation under Grant No. 9984454.